







سیستینوزیس نوعی بیماری ژنتیکی نادر اما خطرناک است که بر اثر تجمع بیش از حد اسیدآمینهای به نام «سیستین» درون سلولها ایجاد میشود. این تجمع منجر به شکلگیری بلورهایی در بافتهای بدن میشود که عملکرد طبیعی اندامها را مختل میکنند. این اختلال معمولاً ابتدا کلیهها و چشمها را درگیر میکند، اما در مراحل پیشرفته میتواند به مغز، عضلات، کبد، تیروئید، پانکراس و بیضهها نیز آسیب برساند.
تشخیص زودهنگام و آغاز درمان در مراحل اولیه نقش مهمی در کنترل روند بیماری دارد و میتواند بهطور چشمگیری کیفیت زندگی مبتلایان را بهبود بخشد.
بررسی اجمالی سیستینوزیس
در بدن افراد سالم، اسیدآمینهها درون سلولها تجزیه و بازیافت میشوند. اما در مبتلایان به سیستینوزیس، به دلیل نقص در پروتئین حملکنندهای به نام سیستینوزین، اسیدآمینه سیستین نمیتواند از درون لیزوزومهای سلول خارج شود. در نتیجه، سیستین تجمع یافته و به تدریج بلور تشکیل میدهد که این امر به تخریب سلولی و آسیب اندامها منجر میشود.
سیستینوزیس یک بیماری مادامالعمر است که معمولاً از دوران کودکی آغاز میشود و بدون درمان مناسب میتواند به نارسایی کلیه و سایر مشکلات جدی منجر شود.
انواع سیستینوزیس
سیستینوزیس بر اساس زمان بروز علائم و شدت آنها به سه نوع اصلی تقسیم میشود:
۱. سیستینوزیس نفروپاتیک (شیرخوارگی یا زودرس)
شایعترین و شدیدترین نوع سیستینوزیس است و حدود ۹۵٪ از بیماران را شامل میشود. در این نوع، نوزادان دچار سندرم فانکونی کلیوی میشوند، وضعیتی که در آن کلیهها قادر به بازجذب مواد مغذی، مواد معدنی و الکترولیتها نیستند.
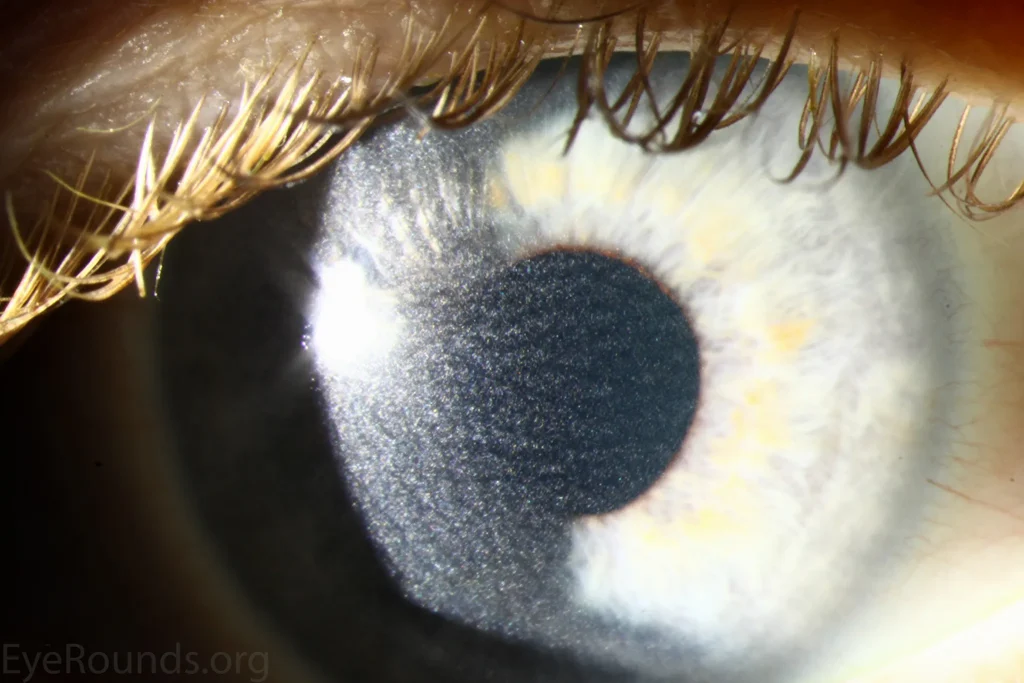
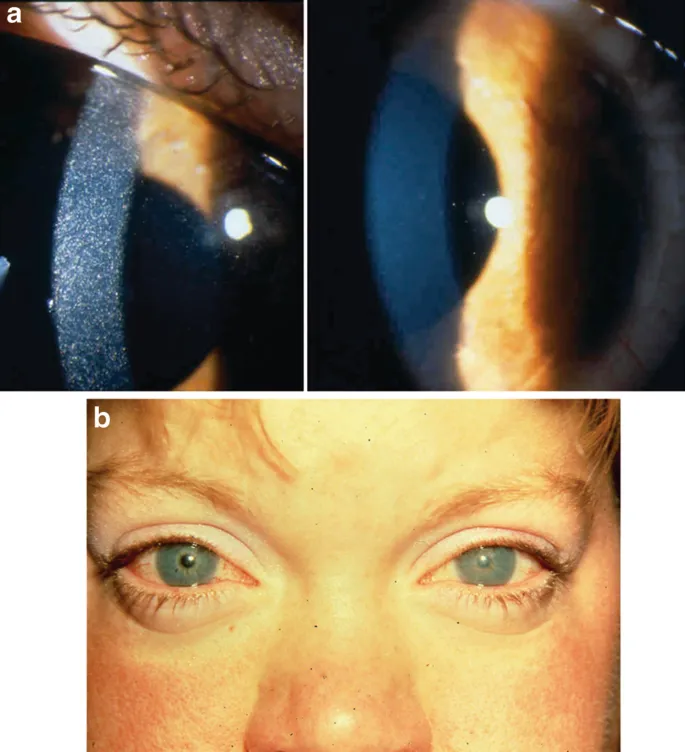
علائم این نوع شامل تأخیر رشد، نرمی استخوانها (راشیتیسم)، تشنگی و ادرار بیش از حد است. در سنین حدود دو سالگی ممکن است بلورهای سیستین در قرنیه چشم ظاهر شوند که باعث حساسیت شدید به نور (فتوفوبیا) و درد چشمی میشود.
در صورت عدم درمان، نارسایی کامل کلیه تا سن ۱۰ سالگی بروز میکند، اما با درمان بهموقع میتوان عملکرد کلیه را تا بزرگسالی حفظ کرد.
۲. سیستینوزیس میانی (نوجوانی یا دیررس)
علائم این نوع مشابه نوع شیرخوارگی است، اما دیرتر، معمولاً در دوران کودکی یا نوجوانی بروز پیدا میکند و شدت آن کمتر است. بیماران ممکن است تا دهه دوم زندگی دچار نارسایی کلیه شوند و در نهایت به پیوند کلیه نیاز پیدا کنند.
۳. سیستینوزیس غیرنفروپاتیک (خوشخیم یا چشمی)
این نوع، تنها قرنیه چشم را درگیر میکند و با تشکیل بلورهای سیستین در چشم همراه است. بیماران معمولاً فقط از حساسیت به نور شکایت دارند و سایر اندامها درگیر نمیشوند. اغلب در سنین بزرگسالی تشخیص داده میشود و پیشرفت آن نسبتاً خفیف است.
علت و شیوه انتقال بیماری
سیستینوزیس به دلیل جهش ژنتیکی در ژن CTNS ایجاد میشود. این ژن دستور ساخت پروتئین سیستینوزین را میدهد که مسئول انتقال سیستین از لیزوزومها به بیرون است.
بیماری به صورت اتوزوم مغلوب به ارث میرسد، یعنی هر دو والد باید حامل نسخه معیوب این ژن باشند تا فرزندشان مبتلا شود. اگر هر دو والد ناقل باشند، احتمال تولد کودک مبتلا ۲۵ درصد است.
شیوع سیستینوزیس
سیستینوزیس از جمله بیماریهای بسیار نادر است و حدود یک مورد در هر ۱۰۰ تا ۲۰۰ هزار تولد در سراسر جهان مشاهده میشود. با این حال، در جوامعی که ازدواجهای فامیلی رایج است، احتمال بروز آن اندکی بیشتر است.
تأثیر بیماری بر بدن
سیستینوزیس نوعی اختلال ذخیرهسازی لیزوزومی است. در این اختلال، مواد زائد و اسیدآمینهها به درستی از سلولها خارج نمیشوند. تجمع بلورهای سیستین باعث آسیب تدریجی به سلولها و در نهایت از کار افتادن اندامهایی مانند کلیه، چشم، تیروئید و پانکراس میشود.
علائم و نشانههای سیستینوزیس
علائم بیماری بر اساس نوع و سن بروز متفاوت است. در نوع نفروپاتیک، نشانهها معمولاً بین ۶ تا ۱۸ ماهگی ظاهر میشوند و شامل موارد زیر هستند:
- تشنگی و ادرار زیاد
- کمآبی بدن و تب
- استفراغ و اختلال الکترولیتی
- راشیتیسم و نرمی استخوان
- تأخیر رشد
- کدورت قرنیه و فتوفوبیا
در انواع دیررستر، علائم خفیفتر اما مشابهاند و ممکن است شامل خستگی، ضعف عضلانی، کوتاهی قد، تأخیر در بلوغ و ناباروری باشند. در نوع چشمی، تنها علامت بیماری حساسیت به نور است و کلیهها سالم باقی میمانند.
روشهای تشخیص سیستینوزیس
پزشکان برای تشخیص بیماری از ترکیبی از معاینه بالینی و آزمایشهای تخصصی استفاده میکنند:
- آزمایش خون: اندازهگیری میزان سیستین در گلبولهای سفید.
- آزمایش ژنتیک: بررسی وجود جهش در ژن CTNS.
- آزمایش ادرار: ارزیابی از دست رفتن مواد مغذی و الکترولیتها.
- معاینه چشم با لامپ شکافدار: شناسایی بلورهای سیستین در قرنیه.
در مادران بارداری که سابقه خانوادگی بیماری دارند، آزمایشهای آمنیوسنتز یا نمونهبرداری از پرزهای جفتی (CVS) میتواند پیش از تولد وجود بیماری را مشخص کند.
درمان سیستینوزیس
درمان قطعی برای سیستینوزیس وجود ندارد، اما درمانهای دارویی میتوانند روند پیشرفت آن را بهطور چشمگیری کند کنند و کیفیت زندگی را افزایش دهند.
درمان دارویی با سیستئامین
داروی اصلی درمان این بیماری، سیستئامین است که سطح سیستین را در سلولها کاهش میدهد. سیستئامین در دو نوع تولید میشود:
- Cystagon™: باید هر ۶ ساعت مصرف شود.
- Procysbi™: دارای پوشش با رهش تأخیری است و هر ۱۲ ساعت مصرف میشود.
این دارو میتواند آسیب کلیوی را به تأخیر بیندازد و به بهبود رشد کودکان کمک کند.
برای درمان بلورهای قرنیه، از قطرههای چشمی سیستئامین مانند Cystaran™ و Cystadrops™ استفاده میشود که به حل بلورها و کاهش فتوفوبیا کمک میکنند.
درمانهای حمایتی و تکمیلی برای سندرم فانکونی کلیوی
کودکانی که به سندرم فانکونی کلیوی مبتلا هستند، ممکن است به مجموعهای از درمانهای حمایتی نیاز داشته باشند تا عملکرد بدنشان حفظ شود و کیفیت زندگیشان بهبود یابد. این اقدامات شامل موارد زیر است:
- تأمین مایعات و الکترولیتها برای جبران کمبودهای بدن
- مصرف داروهایی برای تنظیم سطح فسفات و سایر مواد معدنی
- استفاده از هورمون رشد جهت کمک به افزایش قد
- تزریق انسولین در صورت بروز دیابت
- تجویز تستوسترون برای رفع اختلالات هورمونی در پسران
- بهرهگیری از گفتاردرمانی، مشاوره ژنتیک و در صورت نیاز، استفاده از لوله تغذیه (گاستروستومی) برای تغذیه مناسب
- انجام پیوند کلیه در مراحل پیشرفته بیماری
با وجود این مراقبتها، بسیاری از بیماران در نهایت دچار نارسایی کلیه میشوند و نیازمند پیوند کلیه خواهند بود. خوشبختانه، پیوند کلیه در مبتلایان به سیستینوزیس معمولاً موفقیتآمیز است، زیرا کلیه پیوندی دچار تجمع سیستین نمیشود. با این حال، سایر اندامها ممکن است همچنان تحت تأثیر بیماری باقی بمانند.
ادامه درمان و عوارض جانبی
برای کنترل بیماری، مصرف داروهای کاهنده سیستین مانند سیستئامین باید تا پایان عمر ادامه یابد. این دارو ممکن است با عوارضی مانند طعم و بوی ناخوشایند، تهوع، استفراغ یا سوزش معده همراه باشد. برای کاهش این عوارض، معمولاً از داروهای مهارکننده پمپ پروتون استفاده میشود. همچنین برخی بیماران ممکن است دچار بوی نامطبوع دهان یا تعریق شوند که با رعایت بهداشت فردی قابل مدیریت است.
جمعبندی
سیستینوزیس با وجود نادر بودن، تأثیرات گستردهای بر سلامت فرد دارد و در صورت عدم درمان میتواند منجر به آسیبهای جدی در کلیه و سایر اندامها شود. تشخیص زودهنگام، انجام آزمایشهای ژنتیکی در خانوادههای در معرض خطر، مصرف منظم دارو و مراقبت پزشکی مداوم، در کنار حمایت خانواده، نقش کلیدی در کنترل بیماری و ارتقای کیفیت زندگی بیماران ایفا میکند.